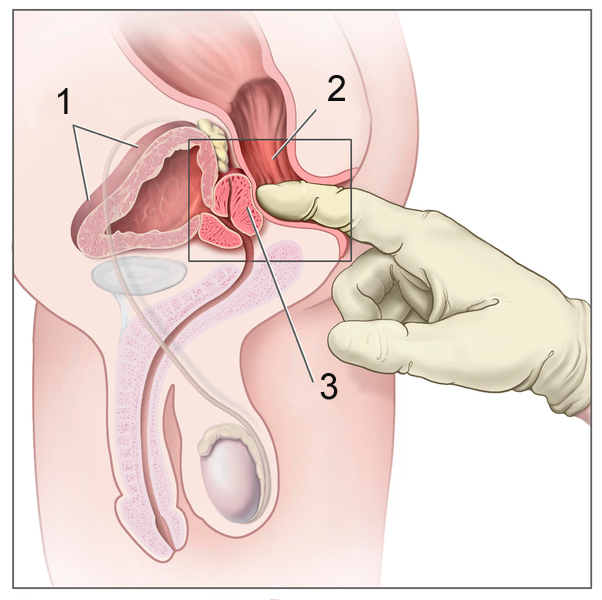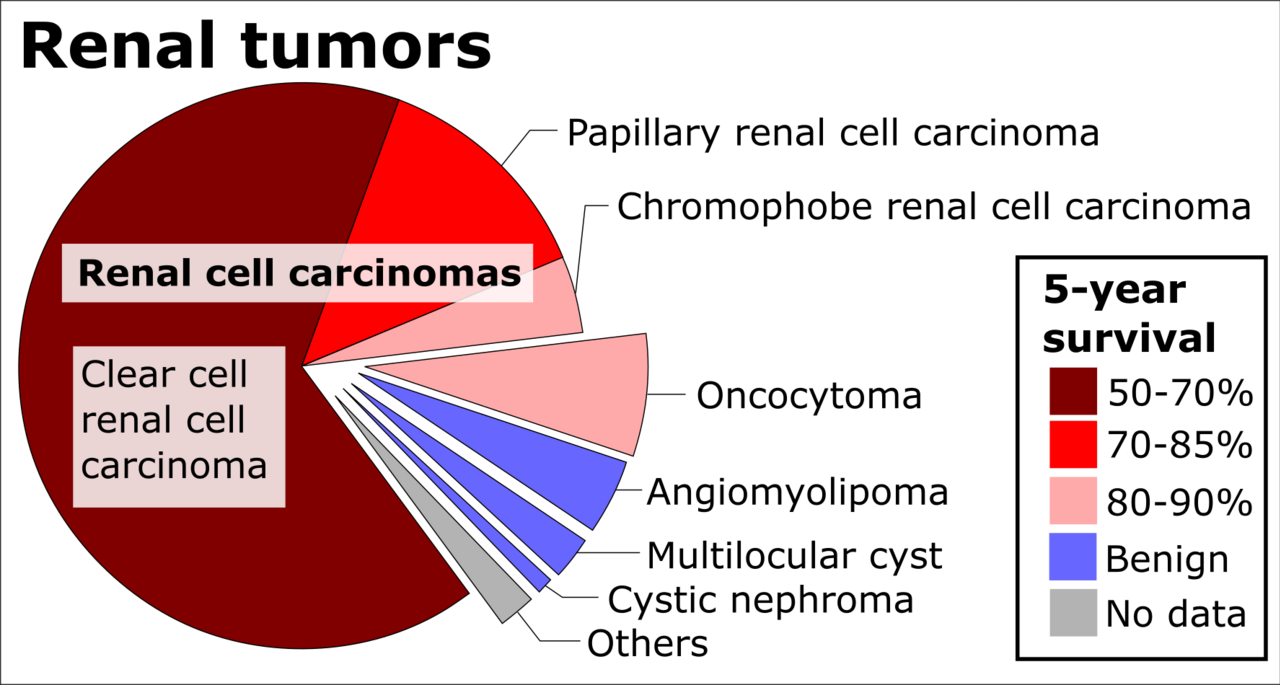
Wikimedia Commons (CC BY-SA)
#001
Principes de l'oncogénétique : hérédité monogénique vs polygénique
- Oncogénétique = étude des prédispositions héréditaires au cancer. Environ 5-10 % des cancers urologiques surviennent dans un contexte de prédisposition génétique identifiable.
- Hérédité monogénique (mendélienne) : mutation d'un seul gène à forte pénétrance, transmission autosomique dominante le plus souvent. Exemples : VHL (chr 3p25), Lynch/MMR (MLH1, MSH2, MSH6, PMS2), BRCA1/2, HOXB13, FH, FLCN, TSC1/TSC2.
- Modèle « two-hit » de Knudson : les patients héréditaires héritent d'un premier allèle muté (1er hit germinal) ; le cancer survient quand le second allèle est inactivé (2e hit somatique) → âge de survenue plus précoce et tumeurs bilatérales/multifocales.
- Hérédité polygénique : combinaison de variants fréquents (SNPs) à faible effet individuel, contribuant au risque global. Scores de risque polygénique (PRS) en cours de validation, notamment pour le cancer de prostate.
- Critères évocateurs d'une prédisposition héréditaire : âge de survenue précoce (< 50 ans), tumeurs bilatérales ou multifocales, histologie particulière (ex. carcinome à cellules claires multifocal bilatéral du rein), antécédents familiaux de cancers du même spectre, association de tumeurs rares.
- L'identification d'une mutation germinale a 3 conséquences : dépistage précoce des apparentés, surveillance spécifique adaptée au syndrome, accès à des thérapeutiques ciblées (PARPi, immunothérapie MSI-H).
⚠️ Ne pas confondre mutation germinale (présente dans toutes les cellules, transmissible) et mutation somatique (acquise dans la tumeur, non transmissible). Seules les mutations germinales relèvent de l'oncogénétique.